
Download

F 0 F = 1 + K S V [ Q ] = 1 + K q τ 0 [ Q ] , (1)
I n K = Δ H R T + Δ S R (2) " Δ G = − R T I n K = Δ H − T Δ S , (3)

ORIGINAL ARTICLE
Interaction behavior between bisphenol AP and pepsin: Insights from density functional theory, and spectroscopic and molecular dynamic simulation
Tianzhu Guan, Ning Li, Ya Gao, Longfei Zhang, Qin Hu, Huaxiang Li, Ming Yang, Lixia Xiao, Lei Yuan*, Zhenquan Yang*
College of Food Science and Engineering, Yangzhou University, Yangzhou, China
Abstract
Overuse of polymer materials has caused increasing direct human exposure to bisphenol AP (BPAP). Through the contaminated environment and food chains, the adverse effects of BPAP on humans and plants induce growing concerns. In this study, the effects of BPAP on pepsin structure changes were exhaustively investigated by multi-spectral methods. Under mimic physiological conditions, BPAP caused a gradient intrinsic fluorescence quenching by inducing microenvironmental changes surrounding residues with the endogenous fluorescence in pepsin. During the ground-state complex formation, the senior structures of pepsin were altered by BPAP addition. Fourier transform infrared spectroscopy and circular dichroism spectroscopy showed that the secondary and tertiary structures of pepsin were changed after the addition of BPAP. Thermodynamic parameter analysis demonstrated that the pepsin and BPAP binding was a spontaneous process that was mainly driven by hydrophobic interaction and van der Waals force. With BPAP as the subject of density functional theory experiments, the energies of the highest occupied molecular orbital, the lowest unoccupied molecular orbital, and the electrostatic potential were calculated to evaluate the electronic distribution of BPAP. Molecular docking experiments displayed that in the specific interaction pattern, hydrogen-bonding between one of the hydroxyl groups and specific amino acids was vital in stabilizing the BPAP−pepsin complex. The root mean square deviation, total hydrogen bonds statistic, and Ramachandran map obtained by molecular dynamic simulations validated the findings and predicted the rationality of the complex structure. This study provides an experimental and theoretical basis for understanding the binding mechanism of bisphenol pollution and pepsin. It also puts forward strategies to strengthen food safety and achieve precise control of environmental contaminants in the food industry.
Key words: bisphenol AP, interaction, molecular dynamic simulation, multi-spectroscopy, pepsin
Corresponding Authors: Lei Yuan and Zhenquan Yang, College of Food Science and Engineering, Yangzhou University, Yangzhou, China. Emails: [email protected] & [email protected]
Received: 2 December 2021; Accepted: 4 January 2022; Published: 11 April 2022
© 2022 Codon Publications
This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0). License (http://creativecommons.org/licenses/by-nc-sa/4.0/)
Introduction
Bisphenol analogues (BPs), ubiquitous endocrine-disrupting chemicals, are extensively applied in the production of thermal paper, epoxy resin, and polycarbonates (Lee et al., 2020; Lin et al., 2017). These synthetic chemicals are also discharged into sewage treatment plants and accumulate in sludge (Zhang et al., 2020). Therefore, the migration of BPs from can coatings, paper products, and different kinds of food materials to food matrices and environmental media is inevitable (Yang et al., 2018). In response to the growing risk of BPA, the migration limit of 600 μg/kg in food-contact applications was set by the European Commission (González et al., 2020). Along with the deepening of BPA toxicological assessment, such as adverse effects on the epigenome of the offspring and metabolic disorders, many countries gradually ban the use of BPA-containing products (Mahmoudi et al., 2018; Wei et al., 2020). As one major alternative of BPA, bisphenol AP (BPAP, 4,4’-(1-phenylethylidene) bisphenol) is used as an indispensable plasticizer and flame retardant in the synthesis of industrial products (Zhang et al., 2013). However, with the wide use of this synthetic product, the exposure levels of humans to BPAP contamination increase significantly. BPAP not only has a similar molecular structure as BPA but also is more hormone-mimicking than typical endocrine disruptors (Stossi et al., 2016). Hence, the potential deleterious effects of BPAP on hormone action and tissue homeostasis require further evaluation (Wang et al., 2015b).
Due to the strong resistance against environmental degradation, BPAP can continuously migrate by different paths into the food and environmental media (Berto-Júnior et al., 2018). Eventually, BPAP can enter the human body through biological enrichment in the food chains. BPAP, after entering the human alimentary system, which contains the digestive proteases in mammal stomachs, can interact with pepsin. During the enzyme-cutting, pepsin can specifically digest certain amino acids and cleave the proteins into peptides or other small-molecular substances. Thus, pepsin is taken as a model protein to study the relationships between conformational change and bioactivities by pollution addition (Li et al., 2015b). Pepsin is composed of two structurally homologous domains: a C-terminal domain and an N-terminal domain, which are almost composed entirely of β-sheets among proteins (Shen et al., 2015). Between the two domains, the active site of pepsin is formed by two aspartate residues, which are important for the enzymatic activity.
The present study elaborates the interaction mode of the pepsin–BPAP complex via multi-spectroscopic methods. Also, molecular docking and molecular modeling calculations were used to predict the binding mechanism of the complex and to further explain the experimental results. We believe that the experimental and theoretical investigations can help to deeply understand the toxicological effect and biological process of pepsin–bisphenol pollutant complexes at the molecular level.
Materials and Methods
Chemicals and reagents
Pepsin and BPAP were purchased from Aladdin Reagent Co. Ltd. (Shanghai, China). The citric acid–sodium citrate buffer was adjusted to pH 2.0 to keep the physiological condition. Stock solutions were prepared with analytical deionized water and stored at 4 °C throughout the experiments.
Ultraviolet–visible (UV-vis) absorption spectra
The UV–vis absorption spectra of pepsin, BPAP, and pepsin–BPAP complex were recorded at 298 K by a UV-3200 spectrophotometer with a path length of 1 cm (Mapada, Shanghai, China). The citrate buffer solution at pH 2.0 was used as the reference solution. The tested complex samples in quartz cuvettes were scanned within 200–500 nm.
Mechanism of fluorescence quenching
The fluorescence spectra were obtained by an F-3200 spectrophotometer (Gangdong Sci. & Tech. Co. Ltd, Tianjin, China) with an entire transparent 1.0-cm quartz cell. The wavelength of the excited spectrum was set at 282 nm with both excitation and emission slits of 10 nm. The emission spectrum was scanned from 300 to 450 nm. A fixed concentration of pepsin (5.0 × 10−5 mol·L−1) and gradient BPAP concentrations (from 0 to 1.0 × 10−4 mol·L−1) were dissolved with the buffer solution. After thorough blending, a thermostat water bath was used to maintain the solution at 298, 304, and 310 K for 30 min.
Synchronous fluorimetry
The synchronous fluorescence spectra of pepsin and pepsin–BPAP complex were measured by the F-3200 spectrophotometer at 298, 304, and 310 K. The wavelength interval (Δλ, λem–λex) was set at 60 nm and recorded in 260–320 nm to investigate the microenvironment changes near Trp and Tyr residues.
Three-dimensional fluorescence spectroscopy
Three-dimensional fluorescence spectra of free pepsin and pepsin–BPAP complex were obtained by the F-3200 spectrophotometer. The initial excitation wavelength was set to 200 nm and the emission range was selected from 200 to 780 nm. And, the excitation and emission slit width were set at 10 nm.
Circular dichroism (CD) spectroscopy
Before the CD spectroscopy measurements, the samples were diluted into the corresponding stable concentrations. To observe the structural change of pepsin after the addition of BPAP, a J-810 Circular Dichroism spectropolarimeter (JASCO Company, Japan) was used to collect the CD spectra from 190 to 260 nm at the scan speed of 100 nm min−1. Finally, the data of spectra were corrected by the contribution from the blank buffer and processed using J-810.
Fourier transform infrared (FT-IR) measurement
FT-IR spectra of pepsin–BPAP and pepsin in 4500–500 cm−1 were recorded at 298 K over 100 scans on a 670-IR + 610-IR spectrometer (Varian, USA) by using a diamond cell anvil method with a nominal range resolution of 4 cm−1.
Quantum chemical investigations of BPAP
For investigating the quantum chemical properties of BPAP, the energy minimization was first produced by the density functional Becke-3-Lee–Yang–Parr/6-31G (d) calculations. Then, the frontier molecular orbital parameters, such as the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO), HOMO-LUMO energy gaps, chemical potential (χ), and chemical hardness (η) were calculated from Gauss View 6.0 and Gaussian 09 (Frisch et al., 2009). Especially, χ is calculated as (ELUMO + EHOMO)/2 and η as the (ELUMO − EHOMO)/2 (Stratmann et al., 1998). The electron excitation analysis of BPAP was conducted on Multiwfn, a quantitative multifunctional wave function analysis program (Tian and Chen, 2012).
Molecular docking
The BPAP and pepsin binding mode were predicted using AutoDock 4.2 and AutoDock Tools (Morris et al., 2009). The initial structure of pepsin (ID: 5PEP) was prepared by removing former ligand, ions, and water molecules in the first step. Default parameters were adopted for the docking simulation based on the Lamarckian genetic algorithm (Wei et al., 2017). The complex was centrally located in an appropriate grid box and the docking results were visualized with PyMol (Delano, 2002; Discovery Studio, 2009).
Molecular dynamics (MD) simulation details
MD simulations of pepsin–BPAP complex were performed using Gromacs 2018 (Peng et al., 2014; Sander et al., 2013). Both ff14SB and GAFF force fields were introduced for the pepsin and BPAP (Daidone et al., 2011). The pepsin–BPAP complex was immersed in a TIP3P water system, in which the sodium ions were added to keep the whole system neutral. Then, MD simulations were performed at 298 K and pressure of 1 bar. After energy minimization, NVT and NPT simulations were performed for equilibration. For the MD simulation, the details were saved every 10 ps with a time step of 2.0 fs. Finally, the MD trajectory data, total hydrogen bonds statistic, and Ramachandran map were recorded and analyzed by Gromacs 2018.
Results and Discussion
Spectrum measurements
The UV–vis absorption spectra of pepsin in the presence of BPAP were recorded to determine the complex formation. The variation of the UV spectrum is always associated with conformational changes of pepsin upon the binding of the small molecule (Zeng et al., 2014). Figure 1 shows the spectra of pepsin, the pepsin–BPAP complex, and BPAP. On the UV spectra, the differences between pepsin and the pepsin–BPAP complex reflect the interaction between pepsin and BPAP. An obvious peak at 275 nm can be observed due to the π − π* transition in the benzene rings of fluorescent residues in pepsin (e.g., phenylalanine (Phe), tyrosine (Tyr), and tryptophane (Trp)) (Shen et al., 2015). After the addition of BPAP, the increased absorbance and blue shift in spectra both confirm the complexation between BPAP and pepsin. Thus, the UV–vis spectra are symptomatic of the conformational changes in the pepsin–BPAP complex. However, more methods for experimental confirmation are needed to further illustrate the specific binding pattern between BPAP and pepsin.
Figure 1. UV-Vis absorption spectra of the pepsin–BPAP system.
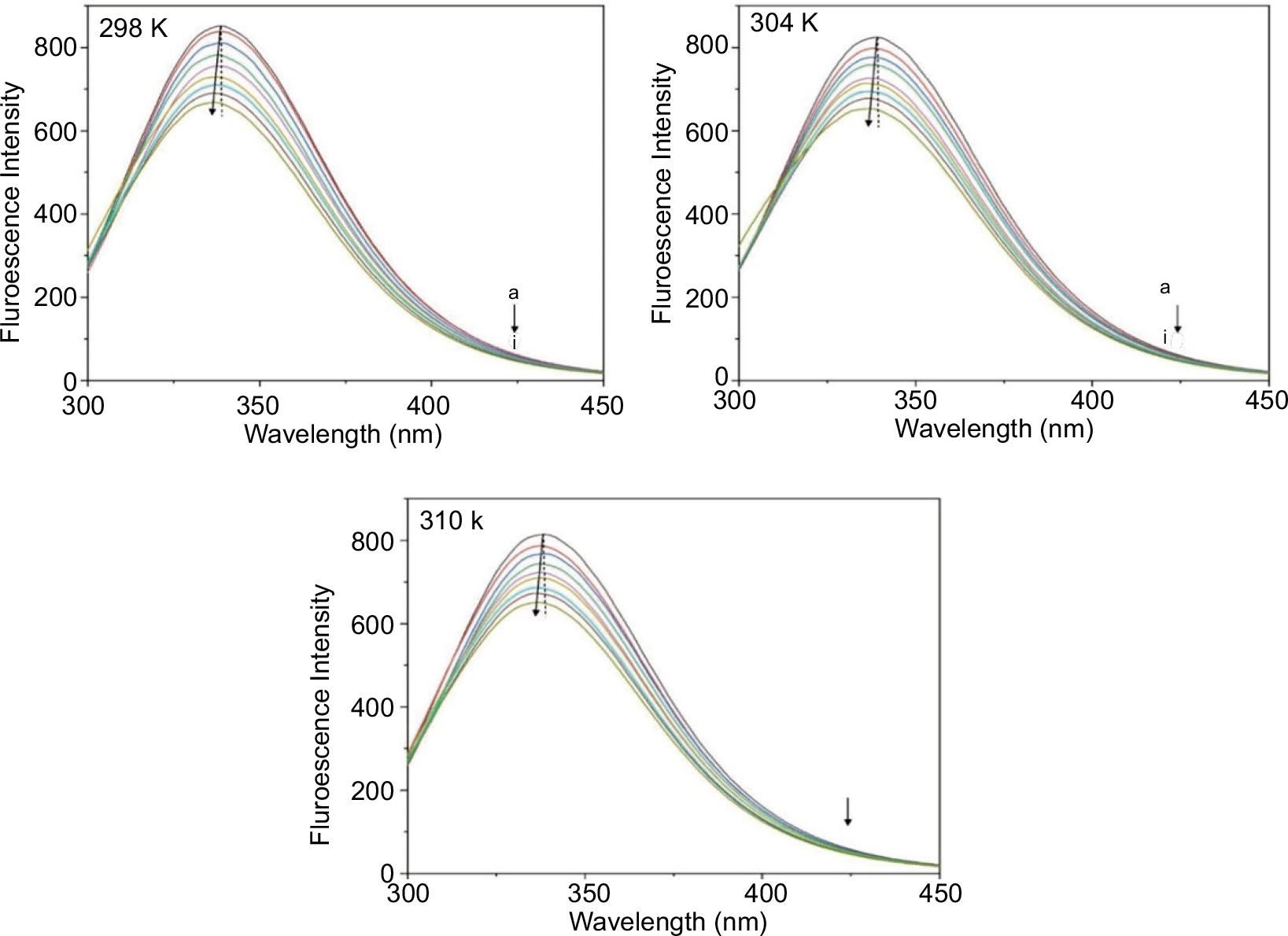
The changes of intrinsic fluorescence can be used to assess the dynamics and microenvironment of pepsin after small-molecule binding. There are multi-Trp residues in pepsin that can be used as a probe due to the contribution of the intrinsic fluorescence. Fluorescence spectra of pepsin and pepsin–BPAP show the maximum emission peak at 345 nm (Figure 2). Upon doping with a concentration gradient of BPAP at 298, 304, and 310 K, the fluorescence intensity of the maximum emission peak decreased regularly, indicating BPAP can bind with pepsin. Also, based on the observations above, it can be inferred that the binding of BPAP altered the polarity environment of the Trp residues at or near the pepsin.
Figure 2. The fluorescence quenching spectra and Sterne–Volmer plots of pepsin and pepsin–BPAP system at 298 K, 304 K, and 310 K. Peaks from top to bottom denote C(BPAP) = (0, 1.25, 2.5, 3.75, 5, 6.25, 7.5, 8.75, 10 × 10−5 mol L−1), C(pepsin) = (5 × 10−5 mol L−1).
Fluorescence quenching mechanism of pepsin
The fluorophore-induced reduction of fluorescence quantum yield is generally called fluorescence quenching, which can be classified into two categories: static quenching and dynamic quenching (Lakowicz, 1999). The association of protein–ligand complexation with static quenching is caused by the ground-state complex formation between the fluorophore and the quencher (Abdelhameed et al., 2017). Dynamic quenching requires a collision between both. To distinguish the two types of quenching, the Stern–Volmer plots can be used to discriminate through the temperature changes:
where F0 and F are the fluorescence intensities of free pepsin and pepsin–BPAP, respectively; KSV is the Stern–Volmer quenching constant, [Q] is the concentration of BPAP, Kq is the quenching rate constant of the biological macromolecule, and τ0 is the fluorescence lifetime in the absence of the quencher (10−8 s).
To elucidate the quenching mechanism, Kq and KSV from Stern–Volmer plots were calculated at 298, 304, and 310 K (Table 1). Within a certain concentration, the good linearity of Stern–Volmer plots represents a single-step quenching process (Figure 2). The KSV at the corresponding systems decreased with the rising temperature. Also, kq is 2.891 × 1011, 2.482 × 1011, and 2.404 × 1011 at 298, 304, and 310 K, respectively, which are larger than the maximum scatter collision quenching constant (2.0 × 1010 mol−1·s−1). Taken together, it can be inferred that the complex formation of pepsin and BPAP was generated by the static mechanism.
Table 1. The quenching rate constants and correlation coefficients of pepsin–BPAP complex.
| T/K | Equations | Ksv (L·mol−1) | Kq (L·mol−1·s−1) | R2 |
|---|---|---|---|---|
| 298 | F0/F = 0.02891[Q] + 1.00601 | 2. 891 × 103 | 2. 891 × 1011 | 0.99368 |
| 303 | F0/F = 0.02482[Q] + 1.00870 | 2.482 × 103 | 2.482 × 1011 | 0.99407 |
| 310 | F0/F = 0.02404[Q] + 1.00721 | 2.404 × 103 | 2.404 × 1011 | 0.99690 |
Binding parameters
To quantitatively understand static quenching, the binding constant (Kb) and the number of binding sites per pepsin molecule (n) were calculated from the plots of log((F0−F)/F) versus log[Q]. The corresponding results of parameters at 298, 304, and 310 K were presented in Table 2, respectively. Kb was 1.6761 × 103, 1.5852 × 103, and 0.6650 × 103 L·mol−1 at 298, 304, and 310 K, respectively, and changed in the order of 298 K304 K310 K, indicating temperature significantly affects the stability of the complex. The rising temperature reduces the stability of the complex, and the interactions in complex formation are exothermic. Notably, n is close to 1, indicating it may be a single binding site of the complex.
Table 2. The binding constants and the number of binding sites for the interactions of BPAP with pepsin at 298 K, 304 K, and 310 K.
| T | Kb (L·mol−1) | n | Fitting equation | R2 |
|---|---|---|---|---|
| 298 | 1.676 × 103 | 0.940 | y = 0.9403 × +3.2243 | 0.996 |
| 303 | 1.585 × 103 | 0.948 | y = 0.9476 × +3.2001 | 0.996 |
| 310 | 0.665 × 103 | 0.861 | y = 0.8609 × +2.8228 | 0.976 |
Pattern of interaction force
In a typical thermodynamic process, free energy change (ΔG), entropy change (ΔS), and enthalpy change (ΔH) can help to understand the essential binding characteristic between pepsin and BPAP. This subsection is aimed to identify the essential noncovalent interactions, such as hydrophobic effect, electrostatic effect, hydrogen bond interactions, and van der Waals force by the van’t Hoff equation:
where K is the binding constant at the corresponding temperature, R is the gas constant, and T is the absolute temperature.
The thermodynamic parameters can be calculated by the thermodynamic laws and summed at different conditions. ΔH < 0 and ΔS > 0 indicate electrostatic interactions and are the major driving forces. ΔH > 0 and ΔS > 0 suggest hydrophobic interactions and are dominant. ΔH < 0 and ΔS < 0 mean that the van der Waals force and hydrogen bond interactions may play a key functional role in the binding reaction. As shown in Table 3, ΔH < 0 and ΔS < 0 in the complexation represent van der Waals force and hydrogen bonding which are the major driving forces, but hydrophobic interaction cannot be excluded at this point. Also, ΔG < 0 in the driving force calculations indicates that the BPAP and pepsin binding process is spontaneous and exothermic.
Table 3. Thermodynamic parameters of the pepsin–BPAP system at different temperatures.
| T/K | ΔH/(kJ·mol−1) | ΔS/(J·mol−1·K−1) | ΔG/(kJ·mol−1) |
|---|---|---|---|
| 298 | −59.168 | −136.824 | −18.394 |
| 303 | −133.369 | −18.623 | |
| 310 | −136. 824 | −16.752 |
Synchronous fluorescence spectra
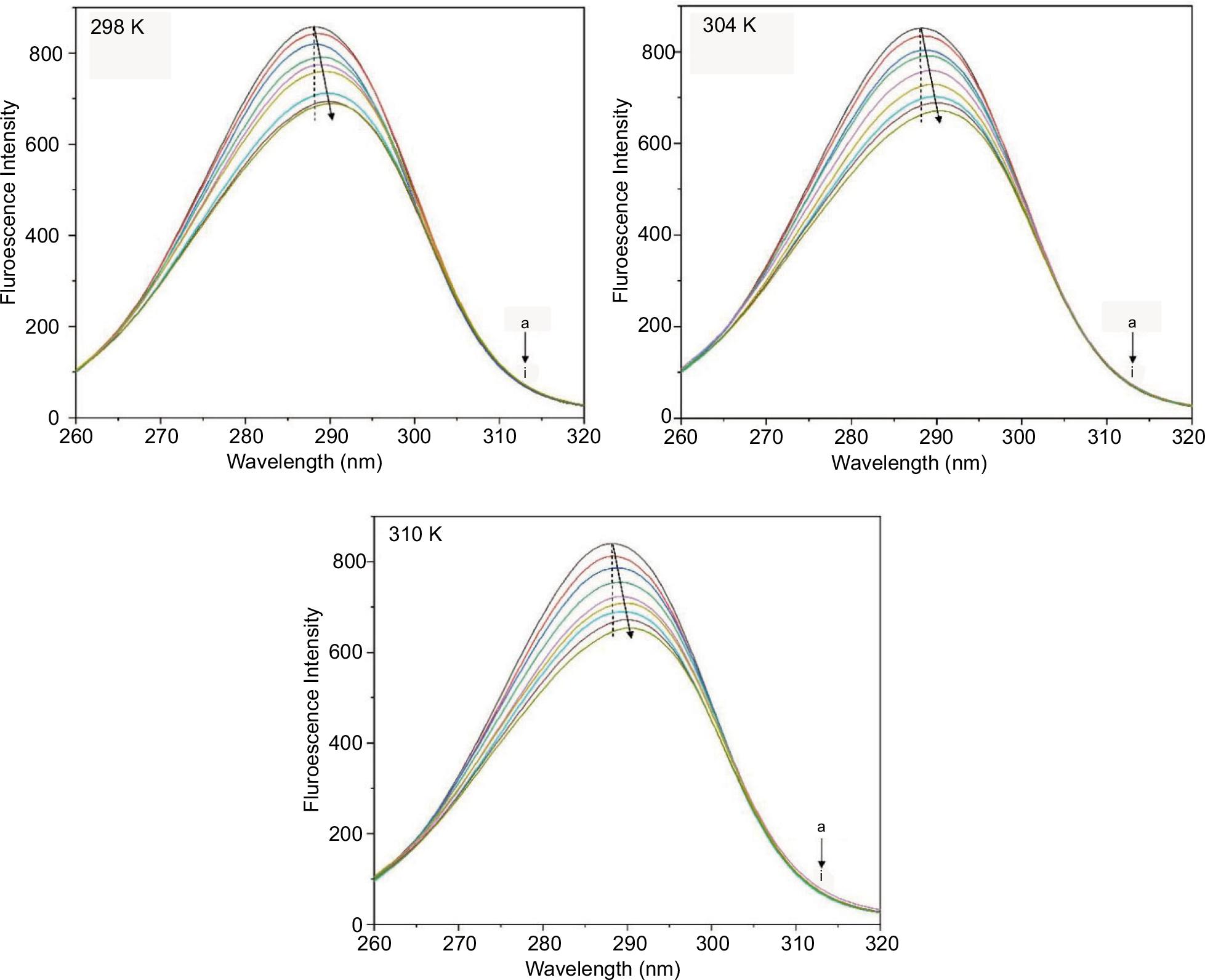
Synchronous fluorescence spectroscopy is one tool to simultaneous measure with the interval (Δλ) between excitation and emission wavelengths fixed at 15 or 60 nm to provide plenty structural information about the chromophore molecules, such as Tyr and Trp residues (Petrov, 1995; Wu et al., 2013). Upon addition of BPAP, the emission spectrum of Trp blue shifts (Figure 3), which may be due to the changes of polarity around the fluorescent amino acid residues. It is further inferred that Trp residues may be previously buried in the nonpolar hydrophobic cavities. After binding with BPAP, the Trp may move into a more hydrophobic surrounding. With the increased hydrophobicity in the micro-environment, the conformation of BPAP and pepsin binding was altered in comparison to the initial structure. However, the changes at Δλ = 15 nm are not significant, indicating BPAP has little effect on the surrounding of Tyr (data not shown). Therefore, the results of synchronous fluorescence measurement indicate that BPAP can strongly affect the microenvironment near Trp.
Figure 3. Synchronous fluorescence spectra of the pepsin–BPAP complex at Δλ = 60 nm at 298 K, 304 K, and 310 K. Peaks from top to bottom denote C(BPAP) = (0, 1.25, 2.5, 3.75, 5, 6.25, 7.5, 8.75, 10 × 10−5 mol L−1), C(pepsin) = (5 × 10−5 mol L−1).
Three-dimensional (3D) fluorescence spectra
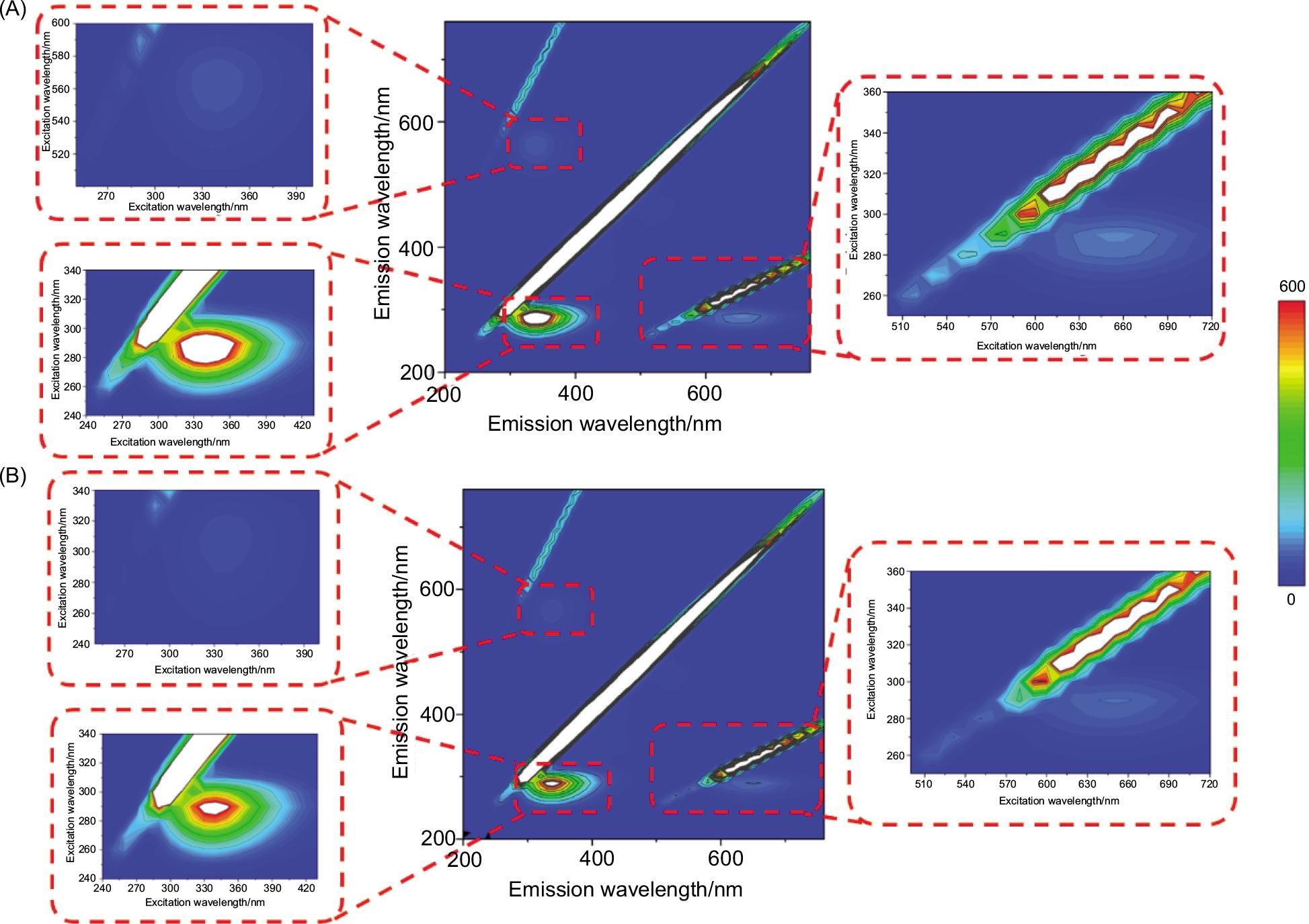
To study the synthetic information of complex formation, 3D fluorescence spectroscopy is a valuable method to get evidence regarding the conformational changes of functional fluorophore (Wang et al., 2019). The 3D array of the fluorescence intensity, excitation, and emission wavelengths of free pepsin and complex pepsin are depicted in Figure 4. Peak 1 represents the Rayleigh scattering peak (λem = λex) and Peak 2 (λem/λex = 336 nm/280 nm) mainly reveals the spectral features of Tyr and Trp residues. It represents the π − π* transitions of the carbonyl groups in the protein backbone. In the presence of BPAP, the large-scale of peak 3 (λem = 2 λex) represents the second-order scattering of the light used for excitation. The increase of peak 3 implies the occurrence of changes in the peptide chain structure of pepsin. In addition, the minor optical changes in peak 4 were observed, but the specific reasons about pepsin and complex need further research. In all, these phenomena reveal that the pepsin and BPAP complex was formed and the interaction of the complex induced a hydrophobic surrounding and conformational alterations.
Figure 4. The three-dimensional fluorescence contour spectra and local enlarged drawing of (A) pepsin and (B) pepsin–BPAP complex.
Conformation investigation
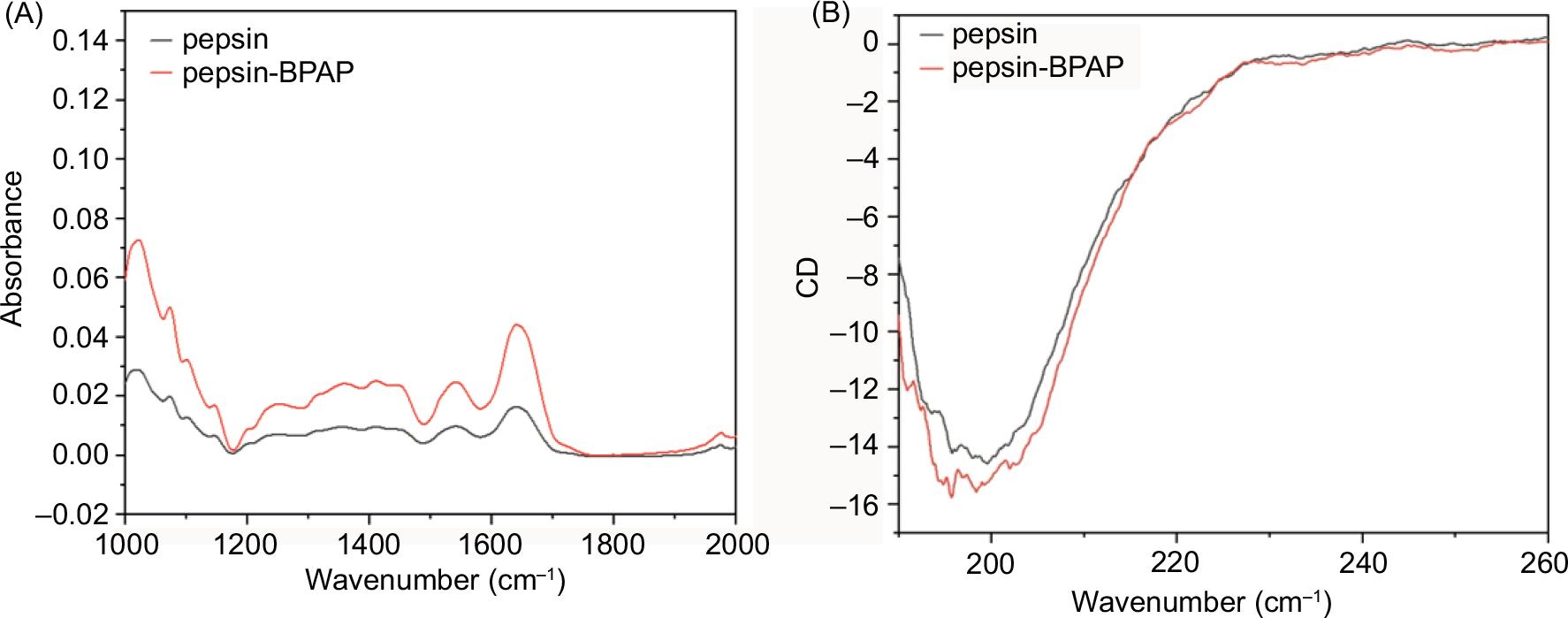
FT-IR is extensively applied to observe the secondary structure changes between free protein and related complex (Gaffney et al., 2009; Yasmin et al., 2019). To further explore the structural changes of pepsin, the changes of the amide I band at 1700–1600 cm−1 and the amide II band at 1600–1500 cm−1 were utilized to interpret secondary structure changes in molecular interaction. Due to the particular sensitivity on typical structure of proteins, the amide I band is assigned to C = O stretching. The influence of BPAP on pepsin structure measured by FT-IR is presented in Figure 5A. On the FT-IR spectrum of the complex, the peak at 1641 cm−1 increases obviously compared with pure pepsin. This increase indicates the occurrence of interactions within the primary amino groups. The increase of the peak at 1540 cm,−1 corresponding to N–H bending and C–N stretching, corroborates the conformational changes upon BPAP binding. These findings support the changes in the secondary structure of pepsin during the BPAP and pepsin binding.
Figure 5. The FT-IR and CD spectra of free pepsin and the pepsin–BPAP complex.
CD measurements
As one well-known and valuable technique, CD spectroscopy is broadly applied to evaluate the conformational changes associated with complex formation. The changes in CD spectra from 190 to 260 nm correspond to the electronic transitions between ground and excited states of proteins (Wang and Zhang, 2014). On the CD spectra of Figure 5B, an obvious negative band at about 200 nm changed, which presents the β-sheet configuration attributed to n−π* transitions. It is also consistent with the literature that the structure of pepsin is predominantly a β-sheet and disordered structure-based conformation (Li et al., 2015a). The presence of BPAP makes the CD spectra in the same shape over the tested range, but the negative molar ellipticity decreases with the BPAP addition. The analysis about the proportion of secondary structure in complexes shows an increase of β-sheet and a decrease of β-turn and random coil structures. This result suggests that after binding with BPAP, the β-turn and random coil structures are partly converted into β-sheet, and that finally a partial change of β-sheet content occurs.
Topology analyses of BPAP
To predict the reactive site of molecules, quantum chemistry calculation can provide insight into the electron structure of tested molecules (Chen et al., 2019; Yan et al., 2020). The calculated HOMO, LUMO, and HOMO–LUMO energy gaps of BPAP after the optimization are presented in Figure 6. Due to the lower ionization potential, the energies of HOMO and HOMO-1 are predictors of electron-donating ability, and are −5.771178 and −5.8346 eV in BPAP, respectively. For the comparison of the electron-acceptive ability of BPAP, the energies of the LUMO and LUMO+1 are –0.260642 and 0.0372 eV, respectively. As an important indicator for chemical reactivity of a molecule, the HOMO–LUMO energy gap corresponding to the charge transfer within the molecule is 5.510526 eV. Based on the calculation results, the chemical potential (χ) and hardness (η) are −3.01591 and 2.755768 eV, respectively, which mean high stability and polarization in the reaction mechanisms. In addition, the correlation between the HOMO–LUMO gap and charge transfer was analyzed by studying the charge transfer within the molecule due to excitation (Rizwana et al., 2018). The charge transfer in an excited state of BPAP is shown in Figure 6. The green and blue isovalue surfaces represent positive values at +0.005 and negative values at −0.005, respectively, corresponding to an increase and a decrease of electron density in an excited state within the BPAP, respectively. The result implies the charge transfer movement after excitation and theoretically underlies the analysis of reaction mechanisms of the protein–ligand interaction.
Figure 6. Topology analyses of the molecular structure of BPAP.
Interaction of pepsin and BPAP
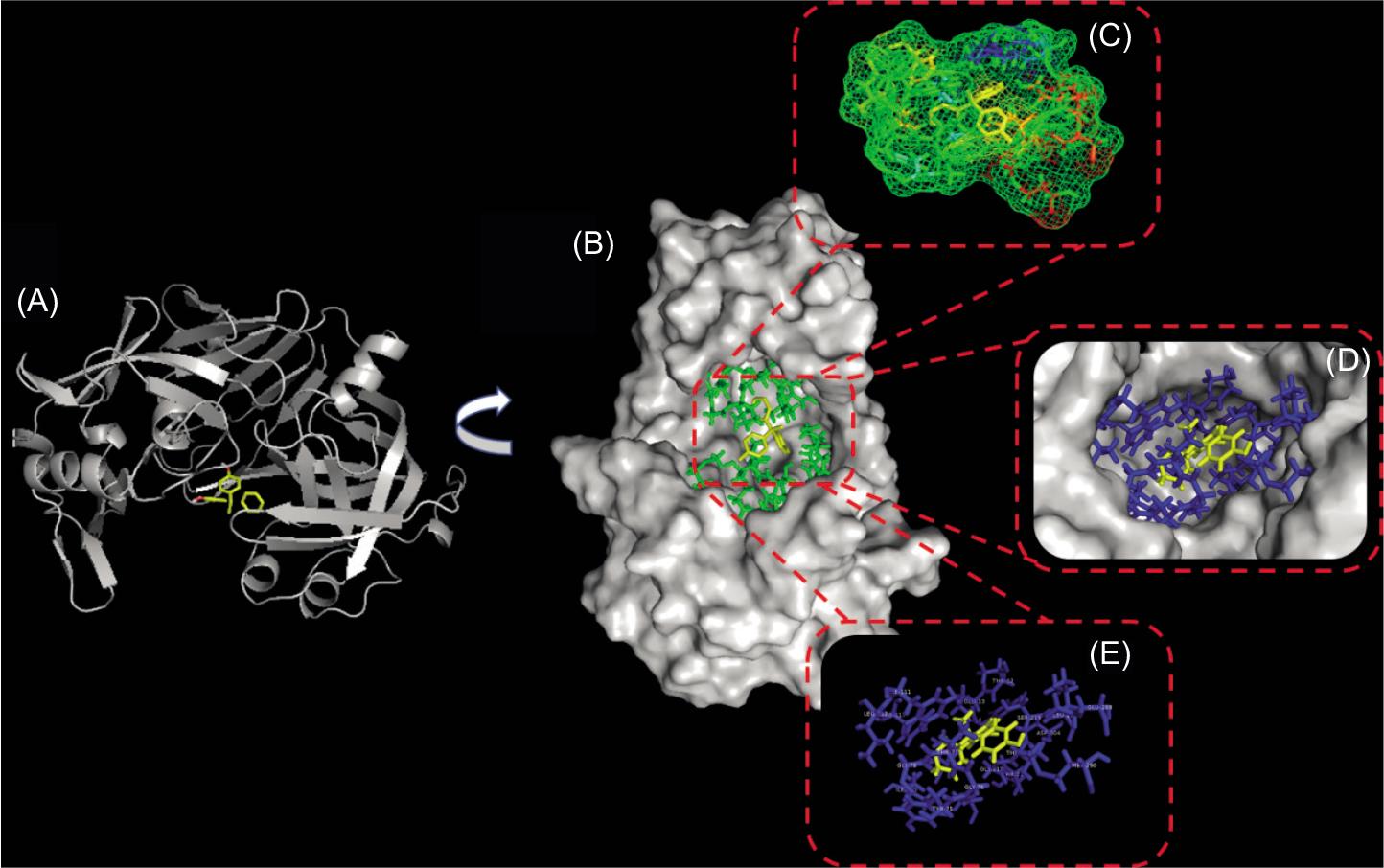
Molecular docking is widely employed to clarify the preferred binding pattern and the lowest interaction energy configuration of the protein–ligand complex (Dumitrașcu et al., 2015; Yu et al., 2021). In terms of macromolecular structures, the central domain of pepsin consists of a six-stranded antiparallel β-sheet that serves as a backbone to the active-site region of molecules (Li et al., 2015b). The stereo view of the BPAP and pepsin binding mode is shown in Figure 7. The docking results demonstrate that BPAP fits into the hydrophobic binding site of pepsin with a structural transformation. Within 5 Å of ligand, several amino acid residues (e.g., Thr-12, Glu-13, Tyr-14, Ile-30, Asp-32, Tyr-75, Gly-76, Gly-78, Phe-111, Leu-112, Phe-117, Ile-120, Ser-219, Leu-220, Met-290, and Asp-304) are located at the binding interface. Docking analysis predicts that one of the two hydroxyl groups on the aromatic rings will interact with Tyr-14, Asp-304, and Thr-216 via hydrogen bonds, which is essentially an electrostatic interaction with strong affinity in nonpolar environments. Therefore, the hydrogen-bonding interactions played an important role in the formation and stability of the pepsin–BPAP complex. Also, π-alkyl and π-anion from the interaction between the aromatic ring of BPAP and the key amino acid residues of pepsin existed at the binding site, which is consistent with the binding energy decomposition diagram of the complex. In addition, due to the hydrophobicity of benzene rings in BPAP, the hydrophobic interactions between BPAP and hydrophobic amino acid residues, and the π−π stacking interactions of BPAP with aromatic residues both played a stabilizing role in the pepsin–BPAP complex formation.
Figure 7. The overview structure of the pepsin-BPAP complex based on molecular docking. (A) Stereo view of the docked conformations of pepsin–BPAP complex. (B) The zoom-on of the binding site of pepsin with BPAP omit electron density map. (C) The binding mode of BPAP in the hydrophobic interactions with pepsin. (D) and (E) Binding geometry of BPAP on pepsin.
Results of MD simulation
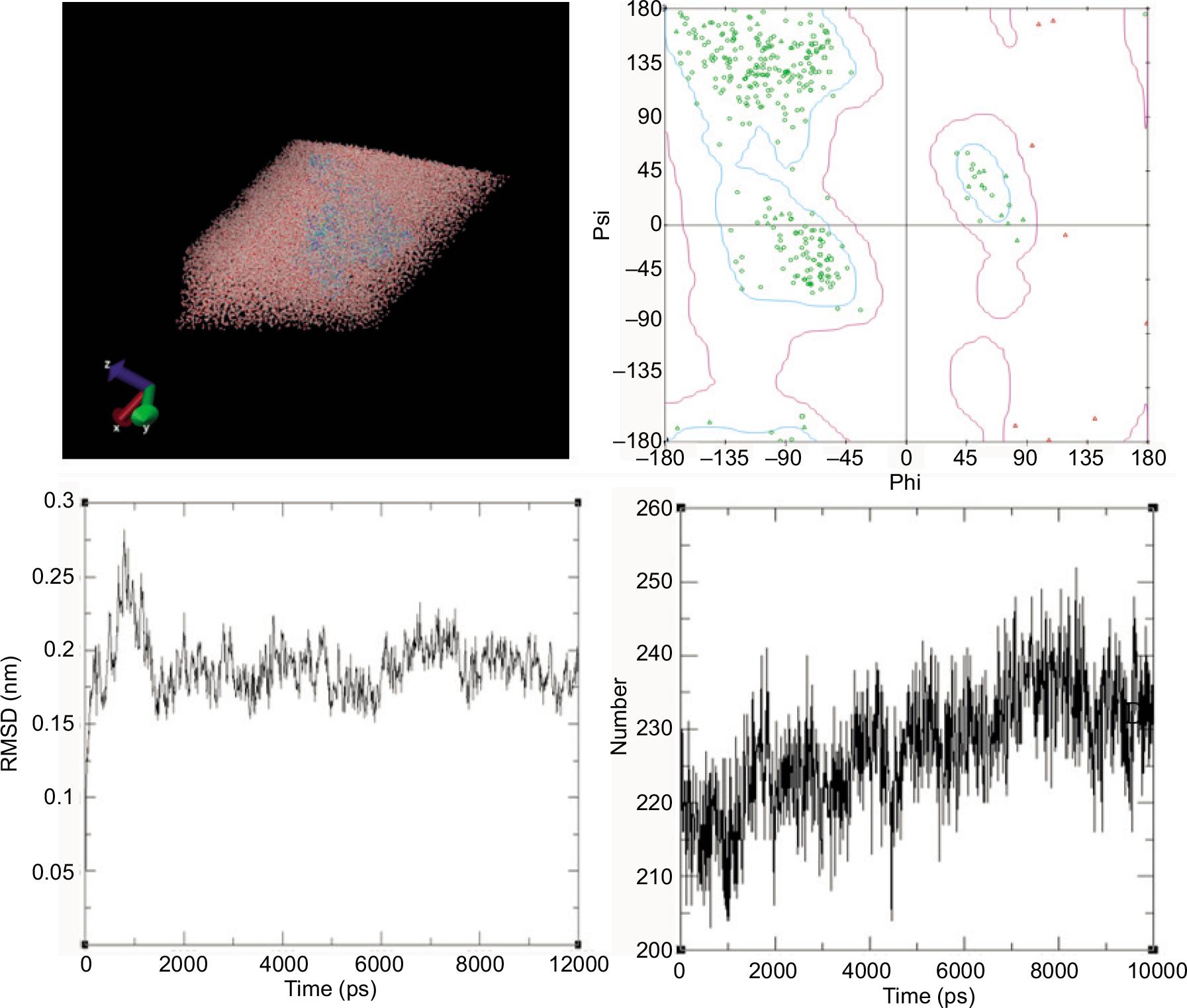
To better understand the BPAP and pepsin binding process, the pose with the minimum binding energy was selected for MD simulation (Shinto, 2013). The initial snapshot of the MD process is shown in Figure 8A. Root mean square deviation (RMSD) is an important indicator to calculate the deviation of Cα backbone atoms from the overlap of the instantaneous structures (Brogi et al., 2020; Wang et al., 2015a; Zhou et al., 2018). A smaller deviation represents a higher spatial equivalent of the compared structures. The violent fluctuations in the plot of RMSD from 0 to 200 ps present much translation and rotation compared with the starting structure (Figure 8C). The RMSD after 200 ps is gradually leveled off, indicating that the interaction of the complex reached its steady-state performance profile.
Figure 8. The overview diagrams of pepsin–BPAP complex molecular dynamics simulation. (A) Relative positions of complex in the water box. (B) Ramachandran plots for the complex. (C) Root mean square deviations for the complex. (D) Distribution of hydrogen bonds of the complex.
Ramachandran map presents the angular deviation in the stereochemistry of protein backbone (Paschek et al., 2011; Yang et al., 2018). Proper distributions of ψand φ angles are quite crucial for the protein conformation. Figure 8B shows the complex with higher percentage of Ramachandran plot exactness, indicating the stereochemical stability. The average quantity of hydrogen bonds during complex formation was calculated through a more detailed analysis (Arnittali et al., 2019). In protein–ligand binding, hydrogen bonds are vital in maintaining the secondary structure, due to the proper distance and angle conditions between the donor and the acceptor. Specifically, the donor and acceptor distance must be shorter than or equal to 0.35 nm and the hydrogen donor–acceptor angle must be less than or equal to 30° (Bashardanesh et al., 2019). The number of hydrogen bonds increased within 0–8500 ps (Figure 8D) and decreased after that. Statistical results showed that the complex reached equilibrium and underwent few conformational changes, which are consistent with the above results.
Conclusions
This study was aimed to explore the mechanism of BPAP and pepsin interaction by using different spectra under a simulated physiological condition. The new pepsin and BPAP complex were formed by a static quenching mode. During the complex formation, BPAP was the quencher to bind with pepsin, and hydrophobic and electrostatic interactions were vital in stabilizing the pepsin–BPAP structure. The effect of BPAP structure was also investigated by the density functional theory, such as energies of HOMO, LUMO, HOMO–LUMO energy gap, chemical potential (χ), and the hardness (η), which provided a theoretical tool to understand the electron mobility during complex formations. Molecular docking and MD simulation showed that hydrogen bonding between the hydroxyl group of BPAP and specific residues occurred in the complex. The calculations of RMSD, Ramachandran plot, and hydrogen bonds are also useful in understanding the pepsin and BPAP interaction. Overall, this work provides important insights into the relationship between BPAP and pepsin. Furthermore, the transport process behavior and the influences of the mixture of the bisphenol pollutions in distribution and elimination will be analyzed in the succeeding work.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 32001661), the Natural Science Foundation of Jiangsu Province, China (Grant Nos. BK20200954 and BK20190890), and the Natural Science Fund for Colleges and Universities in Jiangsu Province, China (Grant No. 20KJB550002). The authors thank T.L. and F.C., for Multiwfn provides and Institute of Theoretical Chemistry (Jilin University, CHN) for the use of Gaussian 09W and GaussView available.
Competing of Interest
The authors declare that there is no conflict of interest.
REFERENCES
Abdelhameed, A.S., Nusrat, S., Ajmal, M.R., Zakariya, S.M., Zaman, M. and Khan, R.H., 2017. A biophysical and computational study unraveling the molecular interaction mechanism of a new Janus kinase inhibitor Tofacitinib with bovine serum albumin. Journal of Molecular Recognition 30: e2601. 10.1002/jmr.2601
Arnittali, M., Rissanou, A.N. and Harmandaris, V., 2019. Structure of biomolecules through molecular dynamics simulations. Procedia Computer Science 156: 69–78. 10.1016/j.procs.2019.08.181
Bashardanesh, Z., Elf, J., Zhang, H. and Spoel, D., 2019. Rotational and translational diffusion of proteins as a function of concentration. ACS Omega 4: 20654–20664. 10.1021/acsomega.9b02835
Berto-Júnior, C., Santos-Silva, A.P., Ferreira, A.C.F., Graceli, J.B., de Carvalho, D.P., Soares, et al. 2018. Unraveling molecular targets of bisphenol A and S in the thyroid gland. Environmental Science and Pollution Research 25: 26916–26926. 10.1007/s11356-018-2419-y
Brogi, S., Sirous, H., Calderone, V. and Chemi, G., 2020. Amyloid β fibril disruption by oleuropein aglycone: long-time molecular dynamics simulation to gain insight into the mechanism of action of this polyphenol from extra virgin olive oil. Food & Function 11: 8122–8132. 10.1039/D0FO01511C
Chen, S., Ouyang, K.-H., Wu, R.-M., Guo, P., Wang, W.-J. and Wang, D., 2019. A sensitive semi-quantitative analysis of patent blue v in drinks with SERS. Quality Assurance and Safety of Crops & Foods 11: 781–788. 10.3920/QAS2019.1639
Daidone, I., Magliano, A., Nola, A.D., Mignogna, G., Clarkson, M.M., Lizzi, A.R., et al. 2011. Conformational study of bovine lactoferricin in membrane-micking conditions by molecular dynamics simulation and circular dichroism. Biometals 24: 259–268. 10.1007/s10534-010-9390-5
Delano, W.L., 2002. PyMOL: an open-source molecular graphics tool. Accelrys Software Inc. Accelrys Draw, Release 4.1, 2013. San Diego: Accelrys Software Inc.
Dumitrașcu, L., Stănciuc, N., Aprodu, I., Ciuciu, A.M., Alexe, P. and Bahrim, G.E., 2015. Monitoring the heat-induced structural changes of alkaline phosphatase by molecular modeling, fluorescence spectroscopy and inactivation kinetics investigations. Journal of Food Science&Technology 52: 6290–6300. 10.1007/s13197-015-1719-1
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. 2009. Gaussian 09W, revision A. 02.
Gaffney, J.S., Marley, N.A. and Jones, D.E., 2009. Fourier transform infrared (FTIR) spectroscopy. American Cancer Society, 101: 157–170. 10.1007/s11120-009-9439-x
González, N., Cunha, S.C., Ferreira, R., Fernandes, J.O. and Domingo, J.L., 2020. Concentrations of nine bisphenol analogues in food purchased from Catalonia (Spain): comparison of canned and non-canned foodstuffs. Food and Chemical Toxicology 136: 110992. 10.1016/j.fct.2019.110992
Lakowicz, J.R., 1999. Quenching of fluorescence. In: Principles of fluorescence spectroscopy. pp. 237–265. 10.1007/978-1-4757-3061-6_8
Lee, S., Eghan, K., Lee, J., Yoo, D., Yoon, S. and Kim, W.-K., 2020. Zebrafish embryonic exposure to BPAP and its relatively weak thyroid hormone-disrupting effects. Toxics 8: 1–15. 10.3390/toxics8040103
Li, S., Peng, Z. and Leblanc, R.M., 2015a. Method to determine protein concentration in the protein–nanoparticle conjugates aqueous solution using circular dichroism spectroscopy. Analytical Chemistry 87: 6455–6459. 10.1021/acs.analchem.5b01451
Li, Z., Li, Z., Yang, L., Xie, Y., Shi, J., Wang, R., et al. 2015b. Investigation of the binding between pepsin and nucleoside analogs by spectroscopy and molecular simulation. Journal of Fluorescence 25: 451–463. 10.1007/s10895-015-1532-2
Lin, Z., Wang, L., Jia, Y., Zhang, Y., Dong, Q. and Huang, C., 2017. A study on environmental bisphenol a pollution in plastics industry areas. Water, Air, & Soil Pollution 228: 98. 10.1007/s11270-017-3277-9
Mahmoudi, A., Hadrich, F., Feki, I., Ghorbel, H., Bouallagui, Z., Marrekchi, R., et al. 2018. Oleuropein and hydroxytyrosol rich extracts from olive leaves attenuate liver injury and lipid metabolism disturbance in bisphenol A-treated rats. Food & Function 9: 3220–3234. 10.1039/C8FO00248G
Morris, G.M., Huey, R., Lindstrom, W., Sanner, M.F., Belew, R.K., Goodsell, D.S., et al. 2009. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry 30: 2785–2791. 10.1002/jcc.21256
Paschek, D., Day, R. and García, A.E., 2011. Influence of water-protein hydrogen bonding on the stability of Trp-cage miniprotein. A comparison between the TIP3P and TIP4P-Ew water models. Physical Chemistry Chemical Physics 13: 19840–19847. 10.1039/c1cp22110h
Peng, W., Ding, F., Jiang, Y.-T., Sun, Y. and Peng, Y.-K., 2014. Evaluation of the biointeraction of colorant flavazin with human serum albumin: insights from multiple spectroscopic studies, in silico docking and molecular dynamics simulation. Food & Function 5: 1203–1217. 10.1039/C3FO60712G
Petrov, E.P., 1995. On the applicability of synchronous fluorescence method to the spectroscopy of systems with dynamic intermolecular interactions. Spectroscopy of biological molecules. 21–22. 10.1007/978-94-011-0371-8_8
Rizwana, F., Christian, J., Muthu, P. and Susan, C., 2018. Molecular docking studies, charge transfer excitation and wave function analyses (ESP, ELF, LOL) on valacyclovir: a potential antiviral drug. Computational Biology and Chemistry 78: 9–17. 10.1016/j.compbiolchem.2018.11.014
Sander, P., Szilárd, P., Roland, S., Per, L., Pär, B., Rossen, A., et al. 2013. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29: 845–854. 10.1093/bioinformatics/btt055
Shen, L., Xu, H., Huang, F., Li, Y., Xiao, H., Yang, Z., et al. 2015. Investigation on interaction between Ligupurpuroside A and pepsin by spectroscopic and docking methods. Spectrochim Acta A Mol Biomol Spectrosc 135: 256–263. 10.1016/j.saa.2014.06.087
Shinto, H., 2013. Molecular dynamics simulation. Shikizai Kyokaishi 86: 380–385. 10.4011/shikizai.86.380
Stossi, F., Dandekar, R.D., Bolt, M.J., Newberg, J.Y. and Mancini, M.A., 2016. High throughput microscopy identifies bisphenol AP, a bisphenol A analog, as a novel AR down-regulator. Oncotarget 7: 16962–16974. 10.18632/oncotarget.7655
Stratmann, R.E., Scuseria, G.E. and Frisch, M.J., 1998. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. Journal of Chemical Physics 109: 8218–8224. 10.1063/1.477483
Tian, L. and Chen, F., 2012. Multiwfn: a multifunctional wavefunction analyzer. Journal of Computational Chemistry 33: 580–592. 10.1002/jcc.22885
Wang, F., Zhang, H., Cuie, G., Sang, S. and Yang, H., 2015a. Mechanism of interaction between sialyltransferase and its inhibitory soyasaponin I. Journal of Food Science and Biotechnology 34: 355–360. 10.1007/s11356-015-4972-y
Wang, L., Liang, T., Ma, J., Sun, L., Yang, C., Meng, L., et al. 2019. Effects of nanoparticle size on the interaction between zinc oxide nanoparticles and bovine serum albumin. Journal of biomolecular Structure & Dynamics 38: 1248–1255. 10.1080/07391102.2019.1596838
Wang, S., Wang, L., Hua, W., Zhou, M., Wang, Q., Zhou, Q., et al. 2015b. Effects of bisphenol A, an environmental endocrine disruptor, on the endogenous hormones of plants. Environmental Science and Pollution Research 22: 17653–17662.
Wang, Y. and Zhang, H., 2014. The effects of bisphenol-S on the structures and activities of trypsin and pepsin. Journal of Agricultural and Food Chemistry 62: 11303–11311. 10.1021/jf504347w
Wei, M., Chai, W.M., Qin, Y., Rui, W. and Peng, Y., 2017. Novel insights into the inhibitory effect and mechanism of proanthocyanidins from pyracantha fortuneana fruit on α-glucosidase. Journal of Food Science 82: 2260–2268. 10.1111/1750-3841.13816
Wei, Y., Han, C., Li, S., Cui, Y., Bao, Y. and Shi, W., 2020. Cuscuta chinensis flavonoids down-regulate the DNA methylation of the H19/Igf2 imprinted control region and estrogen receptor alpha promoter of the testis in bisphenol A exposed mouse offspring. Food & Function 11: 787–798. 10.1039/C9FO02770J
Wu, X., He, W., Wang, W., Luo, X., Cao, H., Lin, L., et al. 2013. Investigation of the interaction between (−)-epigallocatechin-3-gallate with trypsin and α-chymotrypsin. International Journal of Food Science & Technology 48: 2340–2347. 10.1111/ijfs.12223
Yan, B., Sun, Y. and Wei, Y., 2020. Potassium–calcium antagonistic interaction under tomato magnesium deficiency and magnesium fertiliser regulation in solar greenhouse. Quality Assurance and Safety of Crops & Foods 12: 76–86. 10.15586/qas.v12i3.723
Yang, H., Zhang, L., Hou, G. and Liu, C., 2018. Insights into the effect and interaction mechanism of bisphenol S on lipids hydrolysis in sludge through multi-spectra, thermodynamics, and molecule docking analysis. Environmental Science and Pollution Research 25: 7834–7843.
Yasmin, J., Ahmed, M., Lohumi, S., Wakholi, C., Lee, H., Mo, C., et al. 2019. Rapid authentication measurement of cinnamon powder using FT-NIR and FT-IR spectroscopic techniques. Quality Assurance and Safety of Crops & Foods 11: 257–267. 10.3920/QAS2018.1318
Yu, Z., Kan, R., Wu, S., Guo, H., Liu, J., 2021. Xanthine oxidase inhibitory peptides derived from tuna protein: virtual screening, inhibitory activity, and molecular mechanisms. Journal of the Science of Food and Agriculture 101: 1349–1354. 10.1002/jsfa.10745
Zeng, H.J., Qi, T., Yang, R., You, J. and Qu, L.B., 2014. Spectroscopy and molecular docking study on the interaction behavior between nobiletin and pepsin. Journal of Fluorescence 24: 1031–1040. 10.1007/s10895-014-1379-y
Zhang, H., Quan, Q., Zhang, M., Zhang, N. and Wang, Q., 2020. Occurrence of bisphenol A and its alternatives in paired urine and indoor dust from Chinese university students: implications for human exposure. Chemosphere 247: 125987. 10.1016/j.emcon.2021.08.001
Zhang, L., Fang, P., Yang, L., Zhang, J. and Wang, X., 2013. Rapid method for the separation and recovery of endocrine-disrupting compound bisphenol AP from wastewater. Langmuir 29: 3968–3975. 10.1021/la304792m
Zhou, Z., Hu, X., Zhang, G., Wang, R. and Gong, D., 2018. Exploring the binding interaction of the by-product of Maillard reaction 5-hydroxymethyl-2-furaldehyde with calf thymus DNA. Journal of the Science of Food & Agriculture 99: 3192–3202. 10.1002/jsfa.9536