
Download
Determination of fomesafen and quizalofop-p-ethyl in beans using modified QuEChERS-HPLC-DAD
H.C. Tang1, 4, 5*, J.L. Qu1, D.M. Cao1, 2, 3*, W.S. Wang1, Y.H. Lou1
1College of Food Sciences, Heilongjiang Bayi Agricultural University, Daqing 163319, China;
2Key Laboratory of Agro-Products Processing and Quality Safety of Heilongjiang Province, Daqing 163319, China;
3National Coarse Cereals Engineering Research Center, Daqing163319, China;
4Heilongjiang Province Cultivating Collaborative Innovation Center for The Beidah, Heilongjiang, Daqing163319, China;
5Heilongjiang Engineering Research Center for Coarse Cereals Processing and Quality Safety, Daqing 163319, China
Abstract
An improved quick, easy, cheap, effective, rugged, and safe (QuEChERS) method combined with high-performance liquid chromatography (HPLC) coupled to diode array detection was established for the determination of fomesafen and quizalofop-p-ethyl in beans. It is simple and efficient and can be used in most laboratories. This method optimizes four chromatographic conditions, namely, mobile phase, mobile phase ratio, flow rate, and detection wavelength. The effects of extraction agent, extraction dose, extraction time, NaCl amount, and adsorbent on the pretreatment effect were studied. The recoveries were high (92.4–117.8%), and repeatability was good (relative standard deviation [RSD] ranged from 0.35 to 4.36%). The standard solutions ranged from 0.1 to 25 μg/mL. The detection limits of fomesafen and quizalofop-p-ethyl were 0.005 and 0.003 mg/kg, respectively.
Key words: fomesafen, quizalofop-p-ethyl, beans, QuEChERS
*Corresponding Authors: H.C. Tang, [email protected]; D.M. Cao, [email protected]
Received: 04-12-2019| Accepted: 11-06-2020| Published: 10-07-2020
© 2020 Codon Publications
This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
1. Introduction
Beans are important in human nutrition because of their high protein content (20–50%) (Ocloo et al., 2014; Zhao et al., 2019). To obtain high yield, farmers spray their crops with herbicides. Fomesafen and quizalofop-p-ethyl are the most widely used herbicides in the bean fields. Fomesafen is used mainly to control broadleaf weeds as well as some grasses (Sikkema et al., 2009; Zhang et al., 2013), while quizalofop-p-ethyl is used for controlling annual and perennial gramineous weeds in beans (Yadav et al., 2017). According to the database of maximum residue level (MRL) compiled by the U.S. Department of Agriculture (FAS Database, 2019), the MRLs of fomesafen and quizalofop-p-ethyl for bean seed production in the United States are 0.025 and 0.05 mg/kg, respectively.
To date, many methods have been developed for measuring herbicide residues in food, water, and the soil matrix. However, the analysis of these two herbicides in beans has not been reported in the literature. The widely used herbicide detection methods in agricultural products are liquid chromatography (LC)-mass spectrometry (MS) (Stefano et al., 2012), high-performance liquid chromatography (HPLC)-MS (Mantzos et al., 2013), gas chromatography (GC)-MS (Lima et al., 2017), LC-tandem mass spectrometry (MS/MS) (Suganthi et al., 2018), GC-electron capture detection (ECD) (Hu et al., 2010), HPLC-MS/MS (Chen et al., 2014; Guan and Zhang, 2013), capillary electrophoresis (CE)-MS (Safarpour et al., 2004), HPLC-ultraviolet (UV) (Khan et al., 2009), ultra-high performance liquid chromatography (UHPLC)-MS/MS (Chen et al., 2018), and HPLC-photodiode array detection (DAD) (Abbaspour et al., 2019; Liu et al., 2017).
If only sensitivity and selectivity of techniques are considered, MS and tandem MS/MS have been shown to be good choices in the pesticide residue analysis. However, such sensitive detectors are often very specialized and expensive, and many laboratories do not possess them (Lubomirsky et al., 2016). Therefore, we use HPLC-DAD for the analysis of fomesafen and quizalofop-p-ethyl in beans. It is available in standard laboratories for monitoring the herbicide residues of market beans.
A good sample preparation method is important in chromatographic detection. Up till now, some methods of sample preparation for determining pesticide residues have been reported. The most conventionally used extraction techniques for herbicides are liquid–liquid extraction (LLE) (Shi et al., 2018), matrix solid-phase dispersion (MSPD), and solid-phase extraction (SPE) (Climent et al., 2018; Yagüe et al., 2001). SPE has been shown to be effective in a complex matrix. However, the method requires expensive cartridges and sometimes may lead to analyte losses. LLE is time-consuming and requires large amounts of extraction solvents. Recently, the quick, easy, cheap, effective, robust, and safe (QuEChERS) method has been widely used in pesticide detection (Aguilera-Luiz et al., 2001; Hua et al., 2019; Kinsella et al., 2011; Li et al., 2013). Compared with other extraction techniques, the QuEChERS method is safe, simple, economical, effective, efficient, and reliable.
Many improved versions of the QuEChERS method have been developed since its publication. AOAC Official Method 2007.01 uses acetate as a buffer (Lehotay, 2007). CEN Standard Method EN 15662 uses citrate as a buffer. However, due to the influence of different detectors, it was difficult to achieve effective clearance with the QuEChERS method without the MS/MS detection. In particular, when these official QuEChERS methods were used with a low-sensitivity DAD detector, the recoveries were not satisfactory. At present, there are some reports of improved QuEChERS methods to extract pesticides from beans. Abdallah et al. used acetonitrile (ACN) to extract clothianidin from green bean (Abdallah et al., 2019). Wang et al. used the QuEChERS method without the buffer to extract sulfonylurea herbicide from green soybeans (Wang et al., 2015). However, most of these methods work with an expensive MS/MS detector. Considering the economic benefits and the practical applications, it is necessary to establish a cost-effective and more practical method to detect pesticides in beans.
In this work, in order to enhance the extraction of fomesafen and quizalofop-p-ethyl from beans, we improved the QuEChERS preparation approach. We used a mixture of acetic acid and ACN instead of just ACN to extract the two herbicides in beans. We added some NaCl as a substitute for salt mixture. Adding salt before MgSO4 rather than later is beneficial in separating water and organic solvent, averting magnesium sulfate agglomeration immediately upon contact with water, which may also include some unextracted target particles that would result in inaccuracies in the subsequent analysis. Moreover, we used C18 and primary secondary amine (PSA) to absorb, respectively, nonpolar and polar impurities, and optimize extract quality. The improved QuEChERS method can extract herbicides from complex bean matrix quickly, efficiently, and in a cost-effective manner.
2. Materials and methods
Reagents and chemicals
Fomesafen with purity >98.4% and quizalofop-p-ethyl with purity >99% were from Dr. Ehrenstorfer GmbH (Augsburg, Germany). Analytical reagent grade dichloromethane (DCM) was from Liaoning Quanrui Reagent Company Limited (Liaoning, China). Analytical reagent grade acetic acid glacial was purchased from Guangfu Technology Development Company Limited (Tianjin, China). Analytical reagent grade ACN and ethyl acetate (EtOAc) were obtained from Fuyu Fine Chemical Company Limited (Tianjin, China). Chromatographic grade ACN, methanol (MeOH), anhydrous magnesium sulfate, and sorbents for dispersive-SPE, including PSA and C18, were obtained from CNW Technologies Company Limited (Shanghai, China). Pure water was obtained from Hangzhou Wahaha Group Company Limited (Hangzhou, China).
Instrumentation and HPLC conditions
The samples were analyzed using Agilent 1260 Infinity II (Agilent Technologies Incorporated Company, USA). Chromatographic separation was carried out on an Athena C18 column (4.6 × 250 mm, 5 μm, Agilent, USA). A photodiode array detector (DAD) was used. A vortex mixer was purchased from Haimen Qilingbeier Instrument manufacturing Company Limited (Jiangsu, China).
The column temperature was 40°C, and the injection volume was 20 μL. The mobile phase involved the mixture of ACN and acidified water containing 1% acetic acid (70:30, v/v); the flow rate was 1 mL/min, and isocratic elution was used. The detection wavelength was 220 nm. The total run time was 15 min.
Samples and solutions
We use five species of beans, including azuki bean, soybean, mung bean, black soybean, and kidney bean. All the bean seeds were obtained from Daqing farmer’s market (Heilongjiang, China). Unless otherwise specified, the results reported here were obtained by using azuki bean. All five bean species were used only in the experiment reported in the ‘Analysis of bean samples’ section.
The fomesafen and quizalofop-p-ethyl standard stock solutions at a concentration of 100 mg/mL were prepared by adding compound powder to ACN and then storing at 4°C. The samples spiked with fomesafen and quizalofop-p-ethyl (at concentrations of 0.02, 0.2, and 2 mg/kg) were prepared by spiking the mixed standard stock solutions into bean samples.
Sample extraction
The bean sample (5 g) was placed into a 50-mL centrifuge tube, followed by 15 mL of ACN:acetic acid (99:1, v/v) and vortexing for 2 min. Then, 0.3 g of NaCl was mixed in solvent and vortexed for 30 s. Subsequently, 1.5 g of anhydrous MgSO4 was added and vortexed for 30 s, absorbing water to separate ACN from the aqueous matrix. Centrifugation was performed at 4000 rpm for 10 min. The supernatant was evaporated to dryness and then reconstituted with 1 mL of ACN. Sorbent (25 mg) (C18:PSA = 1:1, m/m) was placed into a 2-mL centrifuge tube. The resulting 1 mL solution was transferred into the centrifuge tube, vortexed for 30s, and then centrifuged at 10,000 rpm for 2 min. Finally, the supernatant was taken out, filtered through a 0.22 µm membrane, and then analyzed using HPLC-DAD without further cleanup.
3. Results and discussion
Optimization of HPLC-DAD method
Optimization of mobile phase
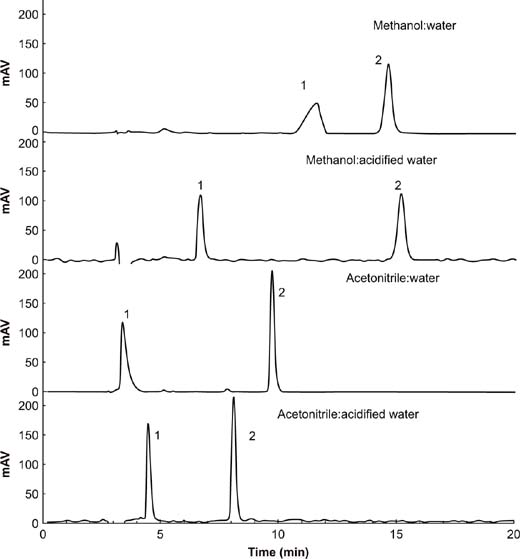
The chromatographic behaviors of four mobile phases, including ACN and MeOH combined with water and acidified water, relative to fomesafen and quizalofop-p-ethyl were investigated for selectivity, sensitivity, and separation effect. The results are shown in Figure 1. ACN had a higher peak area response and shorter retention time compared to MeOH. Replacing water with acidified water improved the peak shape greatly. Therefore, ACN and acidified water were selected for the mobile phase.
Figure 1. Chromatograms of various mobile phases for the determination of fomesafen (1) and quizalofop-p-ethyl (2). Separation conditions: flow rate = 0.8 mL/min; organic solvent: (acidified) water = 80:20; sample volume = 20 μL; λ = 220 nm.
Optimization of mobile phase ratio
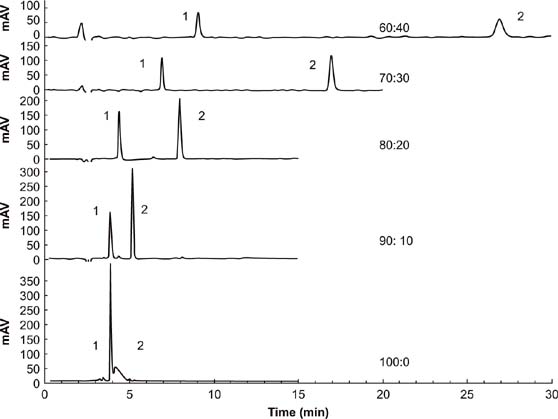
The effect of four ACN/acidified water ratios on the chromatogram separation of fomesafen and quizalofop-p-ethyl is presented in Figure 2. The greater the proportions of ACN, the faster the target analytes were washed out. The reason for this phenomenon was the differential polarity of the column and the solvent. Peak shape was poor at the ratios of 100:0 and 90:10. The distance between the two target peaks was too close in the 90:10 ratio; hence, the separation effect was poor. Sufficient distance should exist between the two target peaks to facilitate the separation of the two objects without being affected by the impurity peaks. The retention time was too long with ACN ratios of less than 70%. According to the peak shapes, the peak separation was better with 70% than 80% ACN. Therefore, the mobile phase composition chosen was 70:30.
Figure 2. Chromatograms of various mobile phase ratios for the determination of fomesafen (1) and quizalofop-p-ethyl (2). Separation conditions: flow rate = 0.8 mL/min; V. sample = 20 μL; λ = 220 nm.
Optimization of flow rate
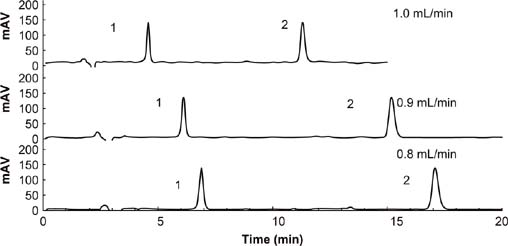
Chromatograms of fomesafen and quizalofop-p-ethyl at three flow rates are shown in Figure 3. The analyte stayed in the column for a short time with a fast flow rate, resulting in a small peak area. Considering the peak area response value and retention time, 1 mL/min was the best choice. At this flow rate, the retention time was within 15 min, and the peak area was 2662 for fomesafen and 4116 for quizalofop-p-ethyl at a concentration of 50 mg/L mixed standard working solution.
Figure 3. Chromatograms of various flows rated for the determination of fomesafen (1) and quizalofop-p-ethyl (2). Separation conditions: acetonitrile:acidified water = 70:30; sample volume = 20 μL; λ = 220 nm.
Optimization of detection wavelength
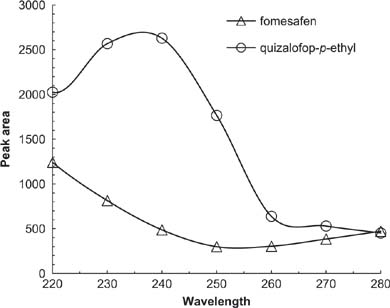
The response results of various wavelength are shown in Figure 4. The peak area response of fomesafen decreased with an increase in wavelength, whereas the peak area response of quizalofop-p-ethyl was always higher. Hence, 220 nm was selected as the detection wavelength at which both fomesafen and quizalofop-p-ethyl were detected sensitively.
Figure 4. Chromatograms of various wavelength. Separation conditions: flow rate = 1 mL/min; acetonitrile:acidified water = 70:30; sample volume = 20 μL.
Optimization of the extraction method
In this work, we adopted an improved QuEChERS method and detected the two herbicides, fomesafen and quizalofop-p-ethyl, in beans by HPLC-DAD. In order to determine the optimal extraction conditions, some parameters affecting the extraction were studied.
Selection of the extraction solvent
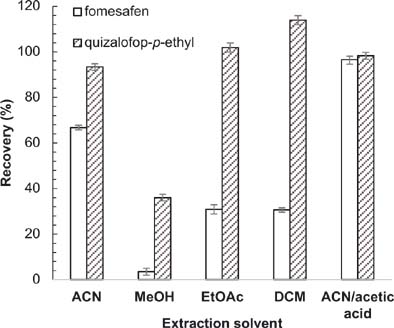
To determine the optimal extractant, five organic solvents, including ACN, MeOH, EtOAc, DCM and ACN:acetic acid (99:1, v/v), were used (Figure 5). Dichloromethane and EtOAc performed efficiently with regard to extraction of quizalofop-p-ethyl but poorly with regard to extraction of fomesafen. Methanol showed the smallest area. Compared with fomesafen, quizalofop-p-ethyl was more sensitive to the acidity and the alkalinity of the solution. Quizalofop-p-ethyl has been reported to decompose easily in an alkaline environment but not in an acidic environment. Therefore, an acidic extraction solution was used. The extraction of quizalofop-p-ethyl by ACN and ACN:acetic acid (99:1, v/v) was similar. In contrast, much better extraction of fomesafen was obtained with ACN:acetic acid (99:1, v/v), compared to ACN. According to the experimental results in Figure 5, the recoveries increased from >60 to >95% after acetic acid was added to the extractant. Thus, ACN:acetic acid was used to extract the analytes.
Figure 5. Extraction effectiveness of various solvents. Conditions: flow rate = 1 mL/min, acetonitrile:acidified water = 70:30; sample volume = 20 μL; extraction solvent volume = 15 mL; extraction time = 2 min; amount of NaCl = 0.3 g; absorbent = 12.5 mg C18 + 12.5 mg PSA.
Determination of acetonitrile:acetic acid volume
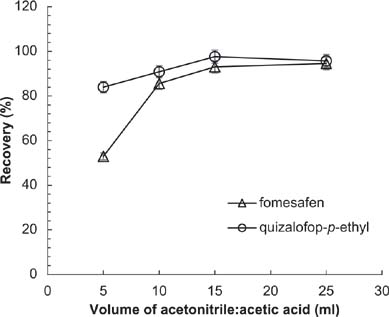
Suitable volume of extraction solvent is needed for good extraction and saving extraction solvent at the same time. As shown in Figure 6, when the volume of extraction solvent increased from 5 to 15 mL, the recovery rate of analyte also increased. When the volume increased above 15 mL, the recovery was unchanged. Therefore, 15 mL was the most appropriate volume of extraction solvent.
Figure 6. Effects of acetonitrile (ACN):acetic acid volume on recovery of analytes. Conditions: flow rate = 1 mL/min; acetonitrile:acidified water = 70:30; sample volume = 20 μL; extraction solvent = ACN:acetic acid (99:1, v/v); extraction time = 2 min; amount of NaCl = 0.3 g; absorbent = 12.5 mg C18 + 12.5 mg PSA.
Determination of extraction time
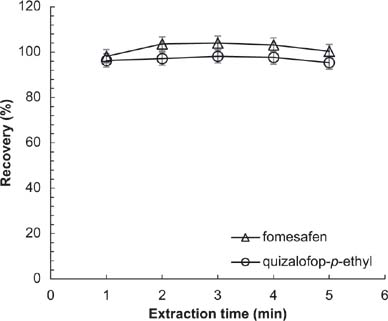
Extraction time should be selected for satisfactory extraction in minimum time. The results (Figure 7) indicated that the recoveries of fomesafen and quizalofop-p-ethyl were approximately constant with extraction time. Given that the extraction time was not an influential variable, and considering sufficient extraction and time saving, 2 min was selected as the best time.
Figure 7. Effects of extraction time on the recovery of analytes. Conditions: flow rate = 1 mL/min; acetonitrile (ACN): acidified water = 70:30; sample volume = 20 μL; extraction solvent = 15 mL ACN:acetic acid (99:1, v/v); amount of NaCl = 0.3 g; absorbent = 12.5 mg C18 + 12.5 mg PSA.
Amount of NaCl
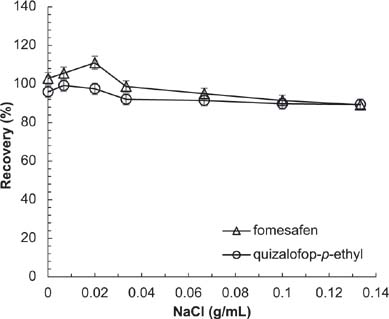
The concentration of salt may influence extraction efficacy of organic solvents. Low concentrations of salt can reduce solubility of herbicides in water and facilitate the transfer of herbicides to the organic solvent. However, high concentrations of salt can cause an increase in viscosity, preventing the herbicide from transferring to the organic solvent. It can be seen in Figure 8 that the extraction effect was best with 0.3 g NaCl per 15 mL of ACN:acetic acid (99:1, v/v).
Figure 8. Effect of NaCl on recovery of analytes. Conditions: flow rate = 1 mL/min; acetonitrile (ACN):acidified water = 70:30; sample volume = 20 μL; extraction solvent = 15 mL ACN:acetic acid (99:1, v/v); extraction time = 2 min; absorbent = 12.5 mg C18 + 12.5 mg PSA.
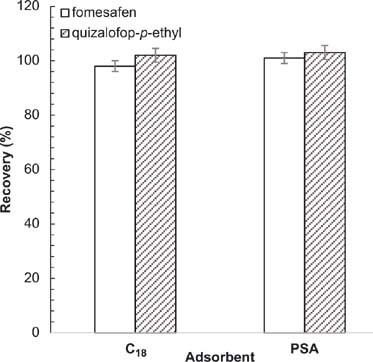
Effect of adsorbent
The effect of adsorbent on purification was studied (Figure 9). The target analytes had a high recovery rate in both C18 and PSA, which were used to absorb polar and nonpolar impurities, respectively, and achieve strong cleanup efficiency. In the purification process, although PSA alone achieved good recoveries, there were impurity peaks that affected the target peak, which influenced the calculation of the target peak response value. After adding C18 at the same time, the impurity peak around the target peak was removed; hence, both PSA and C18 were added.
Figure 9. Effect of adsorbent on recovery of analytes. Conditions: flow rate = 1 mL/min; acetonitrile (ACN):acidified water = 70:30; sample volume 20 μL; extraction solvent = 15 mL ACN:acetic acid (99:1, v/v); extraction time = 2 min; amount of NaCl = 0.3 g; amount of C18 = 25 mg; amount of PSA = 25 mg.
Incomplete water removal in the sample extraction process is not conducive to the water-soluble pesticide entering the ACN layer during extraction. The water contained in the ACN extraction solution during the purification process also reduces the purification effect of the PSA purifier. Therefore, MgSO4 is generally added to the purification process to remove water from the ACN extraction solution. However, the water content was low in beans. NaCl was added to separate the water from ACN, and MgSO4 absorbed water completely during the extraction process. Therefore, MgSO4 was not used in the purification process.
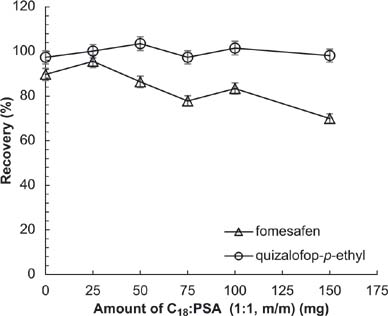
The amount of adsorbent was studied as well (Figure 10). With an increase in the amount of mixed adsorbents C18:PSA (1:1, m/m), there was a decrease in the recovery of fomesafen (suggesting increased adsorption of fomesafen onto mixed adsorbents). Impurities cause unclear chromatograms when adsorbent is not used. Therefore, in order to achieve appropriate purification without affecting the recovery rate, 25 mg of mixed absorbent (C18:PSA) should be used.
Figure 10. Amount of mixed absorbent (C18:PSA, 1:1, m/m). Conditions: flow rate = 1 mL/min; acetonitrile (ACN):acidified water = 70:30; sample volume = 20 μL; extraction solvent = 15 mL ACN:acetic acid (99:1, v/v); extraction time = 2 min; amount of NaCl = 0.3 g.
Evaluation of method performance
Standard curve and limits of quantification
The detection method was verified under optimal conditions. The standard solutions (0.1, 1, 2, 5, and 25 μg/mL) were prepared from the stock solutions, and a standard curve was obtained. The injection quantity was 20 μL, and the chromatographic conditions were the same as above. These curves were used to calculate the recovery rate. The coefficients of determination (R2) (Table 1) were 0.9991 and 0.9999 for fomesafen and quizalofop-p-ethyl, respectively.
Table 1. Standard solution regression equations.
| Analyte | Regression equation | R2 |
|---|---|---|
| Fomesafen | y = 51.15x + 1.24 | 0.9991 |
| Quizalofop-p-ethyl | y = 73.65x + 2.15 | 0.9999 |
The matrix curve (0.02, 0.2, 0.4, 1, and 5 mg/kg) can be obtained by adding five standard working solutions to the blank sample matrix solution, with other conditions being the same as for the standard curve. When the signal-to-noise ratio was 3 (Lubomirsky et al., 2016), the concentration was at the limits of detection (LODs). The LODs for fomesafen and quizalofop-p-ethyl (Table 2) were 0.002 and 0.001 mg/kg, respectively. When the signal-to-noise ratio was 10 (Liu et al., 2010), the limits of quantification (LOQs) were obtained. The LOQs for fomesafen and quizalofop-p-ethyl (Table 2) were estimated to be 0.005 and 0.003 mg/kg, respectively. The LOQs for all analytes were well below the MRL default values mentioned above.
Table 2. Matrix solution regression equation, LODs and LOQs.
| Analyte | Regression equation | R2 | LOD (mg/kg) | LOQ (mg/kg) |
|---|---|---|---|---|
| Fomesafen | y = 55.95x + 9.82 | 0.9992 | 0.002 | 0.005 |
| Quizalofop-p-ethyl | y = 81.10x + 6.98 | 0.9993 | 0.001 | 0.003 |
Matrix effect
Matrix effect (ME) is the effect of compounds other than the target compounds on the response value of the target compounds. In order to improve the accuracy of the determination of target compounds, it is necessary to evaluate the matrix effect. The ME was evaluated by comparing the slope of the matrix curve with those obtained from the solvent curves (Kruve et al., 2008). The results in Table 3 showed that ME was close to 1, meaning that fomesafen and quizalofop-p-ethyl were affected by the matrix effect only to a small extent.
Table 3. Matrix effect of five beans.
| Analyte | Matrix | Regression equation | R2 | Slope ratio (matrix/solvent) |
|---|---|---|---|---|
| Fomesafen | Azuki bean | y = 55.95x + 9.82 | 0.9992 | 1.09 |
| Soybean | y = 54.22x + 5.16 | 0.9995 | 1.06 | |
| Mung bean | y = 56.27x + 8.23 | 0.9993 | 1.10 | |
| Black soybean | y = 53.71x + 4.26 | 0.9998 | 1.05 | |
| Kidney bean | y = 52.17x + 6.72 | 0.9991 | 1.02 | |
| Quizalofop-p-ethyl | Azuki bean | y = 81.10x + 6.98 | 0.9993 | 1.10 |
| Soybean | y = 82.02x + 8.93 | 0.9996 | 1.11 | |
| Mung bean | y = 77.33x + 5.83 | 0.9991 | 1.05 | |
| Black soybean | y = 75.86x + 9.31 | 0.9998 | 1.03 | |
| Kidney bean | y = 80.28x + 6.35 | 0.9992 | 1.09 |
Accuracy and precision
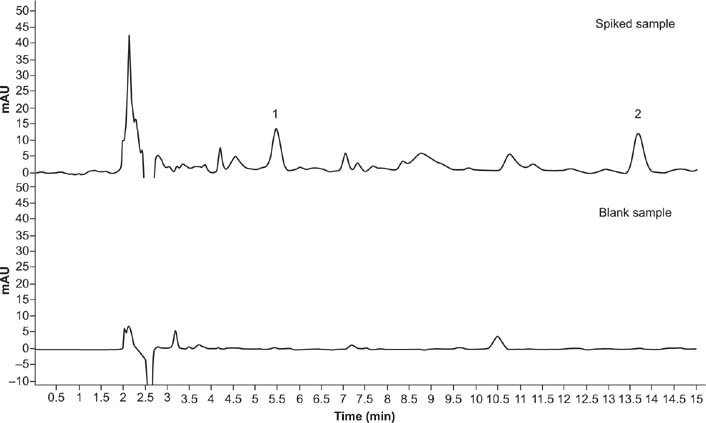
Figure 11 shows the chromatogram of the blank sample and the spiked sample. The accuracy and the precision of the experimental method are generally expressed by determining the spiked recovery rates of the samples. Intra-day precision was determined on red azuki bean samples spiked with concentrations of 0.02, 0.2, and 2 mg/kg five times a day. For inter-day precision, three azuki bean samples spiked at concentrations specified above were measured once a day for five consecutive days. The precision of the method was obtained by calculating RSD (Table 4). The recovery rates ranged from 97.1 to 116.2% and RSD from 0.17 to 3.79%. The accuracy was therefore adequate for meeting the requirements.
Figure 11. Chromatograms of spiked and blank samples (1 is fomesafen and 2 is quizalofop-p-ethyl). Conditions: flow rate = 1 mL/min; acetonitrile (ACN):acidified water = 70:30; sample volume = 20 μL; extraction solvent = 15 mL ACN:acetic acid (99:1, v/v); extraction time = 2 min; amount of NaCl = 0.3 g; absorbent = 12.5 mg C18 + 12.5 mg PSA.
Table 4. Recoveries and RSDs for fomesafen and quizalofop-p-ethyl in azuki bean.
| Added (mg/kg) | Inter-day (n = 5) | Intra-day (n = 5) | |||
|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | ||
| Fomesafen | 0.02 | 99.9 | 3.59 | 97.1 | 3.05 |
| 0.2 | 110.9 | 0.52 | 103.9 | 3.38 | |
| 2 | 116.2 | 0.82 | 112.9 | 1.52 | |
| Quizalofop-p-ethyl | 0.02 | 102.2 | 3.17 | 101.5 | 3.79 |
| 0.2 | 112.9 | 0.78 | 112.7 | 2.14 | |
| 2 | 115.0 | 0.17 | 115.7 | 2.27 | |
Analysis of bean samples
The feasibility of the method was evaluated by measuring the recovery rate of market bean samples (azuki bean, soybean, mung bean, black bean, and kidney bean) spiked with concentrations of 0.02, 0.2, and 2 mg/kg. As shown in Table 5, the recoveries of analytes ranged from 92.4 to 117.8% and RSD from 0.35 to 4.36%. The experimental results showed that the method was applicable and effective. The nonspiked market samples of these five bean species were tested by the same method, and no pesticide residues were found.
Table 5. Analytical results for fomesafen and quizalofop-p-ethyl in spiked samples of five bean species.
| Sample | Added (mg/kg) | Fomesafen | Quizalofop-p-ethyl | ||
|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | ||
| Azuki bean | 0.02 | 99.8 | 2.91 | 101.5 | 3.45 |
| 0.2 | 117.8 | 1.03 | 112.7 | 2.55 | |
| 2 | 114.0 | 0.35 | 115.5 | 0.72 | |
| Soybean | 0.02 | 97.9 | 2.11 | 101.4 | 3.48 |
| 0.2 | 109.9 | 4.19 | 102.3 | 2.47 | |
| 2 | 105.7 | 0.80 | 113.2 | 1.27 | |
| Mung bean | 0.02 | 92.4 | 3.24 | 103.9 | 2.91 |
| 0.2 | 95.5 | 2.87 | 111.5 | 1.85 | |
| 2 | 109.9 | 2.25 | 104.6 | 0.75 | |
| Black soybean | 0.02 | 95.2 | 4.07 | 101.5 | 3.73 |
| 0.2 | 99.9 | 3.72 | 109.9 | 2.79 | |
| 2 | 109.4 | 3.33 | 102.1 | 1.85 | |
| Kidney bean | 0.02 | 94.9 | 4.36 | 98.1 | 3.36 |
| 0.2 | 96.7 | 2.42 | 105.6 | 2.53 | |
| 2 | 106.8 | 2.58 | 110.0 | 2.63 | |
Comparison of the present method with the reference method
The comparison between the detection method, which used ACN:acetic acid as the extractant, and the reference method, which used ACN and water, is shown in Table 6. In the reference method, the amount of C18 was twice that of what we used, and the amount of PSA was 16 times that of what we used. Although increasing the amount of PSA can achieve a better purification effect, PSA absorbs fomesafen. Hence, we could not use too much PSA in our method. Compared with the HPLC-MS/MS method in the reference, the HPLC-DAD method established in this study is cost-effective and is more convenient for the detection of herbicide residues in beans in a nonspecialized laboratory.
Table 6. Comparison of the present method with the reference method.
| Method | HPLC-MS/MS | HPLC-DAD |
|---|---|---|
| Herbicides | Benazolin-ethyl and quizalofop-p-ethyl | Fomesafen and quizalofop-p-ethyl |
| Recoveries | 74.7–110.0% | 92.4–117.8% |
| Extraction Solvent | 10 mL acetonitrile and 5 mL water | 15 mL acetonitrile:acetic acid (99:1, v/v) |
| Adsorbent | 200 mg PSA + 50 mg C18 | 12.5 mg C18 + 12.5 mg PSA |
| Cost | High | Low |
| References | Guan and Zhang, 2013 | This work |
4. Conclusions
A modified QuEChERS method followed by HPLC-DAD analysis was established to detect the herbicides, fomesafen and quizalofop-p-ethyl, in various bean species. The recovery, RSD, LODs, and LOQs of the method all meet the requirements. This method is suitable for most laboratories, with fast preparation and detection speed, low cost, and small consumption of reagents. The detection method proposed in this paper has the potential to be used successfully in the practical detection of herbicides in bean samples.
Acknowledgements
This study was funded by (1) the Department of Education, Heilongjiang Province (grant number [2018] No. 4), (20 the National Key R&D Program of China (2018YFE0206300 and 2018YFE0206300-10), and (3) the Heilongjiang Bayi Agricultural University (TDJH201806).
Conflicts of interest
The authors report no conflicts of interest.
REFERENCES
Abbaspour, M., Farajzadeh, M.A., Sorouraddin, S.M. and Mohebbi, A., 2019. Monitoring of nine pesticides in different cereal flour samples with high performance liquid chromatography-diode array detection. Analytical Methods 11: 4022–4033. 10.1039/C9AY00935C
Abdallah, O.I., Abd El-Hamid, R.M. and Raheem, E.H., 2019. Clothianidin residues in green bean, pepper and watermelon crops and dietary exposure evaluation based on dispersive liquid-liquid microextraction and LC–MS/MS. Journal of Consumer Protection and Food Safety 14: 293–300. 10.1007/s00003-019-01218-4
Aguilera-Luiz, M.M., Plaza-Bolaños, P., Romero-González, R., Vidal, J.M. and Frenich, A.G., 2011. Comparison of the efficiency of different extraction methods for the simultaneous determination of mycotoxins and pesticides in milk samples by ultra-high-performance liquid chromatography-tandem mass spectrometry. Analytical and Bioanalytical Chemistry 399: 2863–2875. 10.1007/s00216-011-4670-7
Chen, L., Chen, J.F., Guo, Y., Li, J.R., Yang, Y.Q., Xu, L.J. and Fu, F.F., 2014. Study on the simultaneous determination of seven benzoylurea pesticides in Oolong tea and their leaching characteristics during infusing process by HPLC–MS/MS. Food Chemistry 143: 405–410. 10.1016/j.foodchem.2013.08.027
Chen, Y., Guo, M., Liu, X., Xu, J., Dong, F., Wu, X., Li, B. and Zheng, Y., 2018. Determination and dissipation of afidopyropen and its metabolite in wheat and soil using QuEChERS–UHPLC–MS/MS. Journal of Separation Science 41: 1674–1681. 10.1002/jssc.201700773
Climent, M.J., Sánchez-Martín, M.J., Rodríguez-Cruz, M.S., Pedreros, P., Urrutia, R. and Herrero-Hernández, E., 2018. Determination of pesticides in river surface waters of Central Chile using SPE-GC-MS multi-residue method. Journal of the Chilean Chemical Society 63: 4023–4031. 10.4067/s0717-97072018000204023
FAS Maximum Residue Limits (MRL) Database, 2019. Available at: https://www.fas.usda.gov/maximum-residue-limits-mrl-database.
Guan, W. and Zhang, H., 2013. Determination and study on residue and dissipation of benazolin-ethyl and quizalofop-p-ethyl in rape and soil. International Journal of Environmental Analytical Chemistry 93: 679–691. 10.1080/03067319.2012.684047
Hu, J., Deng, Z., Liu, C. and Zheng, Z., 2010. Simultaneous analysis of herbicide metribuzin and quizalofop-p-ethyl residues in potato and soil by GC-ECD. Chromatographia 72: 701–706. 10.1365/s10337-010-1717-4
Hua, J., Fayyaz, A., Song, H., Tufail, M.R. and Gai, Y., 2019. Development of a method Sin-QuEChERS for the determination of multiple pesticide residues in oilseed samples. Quality Assurance and Safety of Crops & Foods 11: 511–516. 10.3920/QAS2019.1557
Khan, B.A., Farid, A., Asi, M.R., Shah, H. and Badshah, A.K., 2009. Determination of residues of trichlorfon and dimethoate on guava using HPLC. Food Chemistry 114: 286–288. 10.1016/j.foodchem.2008.08.092
Kinsella, B., Byrne, P., Cantwell, H., McCormack, M., Furey, A. and Danaher, M., 2011. Determination of the new anthelmintic monepantel and its sulfone metabolite in milk and muscle using a UHPLC–MS/MS and QuEChERS method. Journal of Chromatography B 879: 3707–3713. 10.1016/j.jchromb.2011.10.011
Kruve, A., Künnapas, A., Herodes, K. and Leito, I., 2008. Matrix effects in pesticide multi-residue analysis by liquid chromatography–mass spectrometry. Journal of Chromatography A 1187: 58–66. 10.1016/j.chroma.2008.01.077
Lehotay, S.J., 2007. Determination of pesticide residues in foods by acetonitrile extraction and partitioning with magnesium sulfate: collaborative study. Journal of Aoac International 90: 485–520. 10.1093/jaoac/90.2.1SUP
Li, N., Lei, L., Nian, L., Zhang, R., Wu, S., Ren, R., Wang, Y., Zhang, H. and Yu, A., 2013. A modified QuEChERS method for the determination of some herbicides in yogurt and milk by high performance liquid chromatography. Talanta 105: 219–228. 10.1016/j.talanta.2012.11.057
Lima, V.G., Campos, V.P., Santana, T.C., Santana, F.O. and Costa, T.A., 2017. Determination of agrochemical multi-residues in grapes. Identification and confirmation by gas chromatography-mass spectrometry. Analytical Methods 9: 5880–5889. 10.1039/C7AY01448A
Liu, S., Zheng, Z., Wei, F., Ren, Y., Gui, W., Wu, H. and Zhu, G., 2010. Simultaneous determination of seven neonicotinoid pesticide residues in food by ultraperformance liquid chromatography tandem mass spectrometry. Journal of Agricultural and Food Chemistry 58: 271–278. 10.1021/jf904045j
Liu, Y.H., Chen, P.S. and Huang, S.D., 2017. Determination of diphenylether herbicides in water samples using dispersive liquid–liquid microextraction combined with high-performance liquid chromatography. Journal of AOAC International 100: 212–217. 10.5740/jaoacint.16-0078
Lubomirsky, E., Padro, J.M. and Reta, M.R., 2016. Development of a dispersive liquid-liquid microextraction technique for the analysis of aryloxyphenoxy-propionate herbicides in soy-based foods. Microchemical Journal 129: 63–70. 10.1016/j.microc.2016.06.015
Mantzos, N., Karakitsou, A., Zioris, I., Leneti, E. and Konstantinou, I., 2013. QuEChERS and solid phase extraction methods for the determination of energy crop pesticides in soil, plant and runoff water matrices. International Journal of Environmental Analytical Chemistry 93: 1566–1584. 10.1080/03067319.2013.803282
Ocloo, F.C., Okyere, A.A. and Asare, I.K., 2014. Physicochemical, functional and pasting properties of flour produced from gamma irradiated tiger nut (Cyperus esculentus L.). Radiation Physics and Chemistry 103: 9–15. 10.1016/j.radphyschem.2014.05.010
Safarpour, H., Asiaie, R. and Katz, S., 2004. Quantitative analysis of imazamox herbicide in environmental water samples by capillary electrophoresis electrospray ionization mass spectrometry. Journal of Chromatography A 1036: 217–222. 10.1016/j.chroma.2004.03.002
Shi, X.Z., Sun, A.L., Wang, Q.H., Hengel, M. and Shibamoto, T., 2018. Rapid multi-residue analysis of herbicides with endocrine-disrupting properties in environmental water samples using ultrasound-assisted dispersive liquid–liquid microextraction and gas chromatography–mass spectrometry. Chromatographia 81: 1071–1083. 10.1007/s10337-018-3530-4
Sikkema, P.H., Shropshire, C. and Soltani, N., 2009. Response of dry bean to pre-plant incorporated and pre-emergence applications of S-metolachlor and fomesafen. Crop Protection 28: 744–748. 10.1016/j.cropro.2009.05.011
Stefano, V.D., Avellone, G., Bongiorno, D., Cunsolo, V., Muccilli, V., Sforza, S., Dossenac, A., Drahosd, L. and Vékey, K., 2012. Applications of liquid chromatography–mass spectrometry for food analysis. Journal of Chromatography A 1259: 74–85. 10.1016/j.chroma.2012.04.023
Suganthi, A., Nikita, S.A., Kousika, J., Bhuvaneswari, K. and Sridharan, S., 2018. Determination of thiamethoxam residues in banana stem and fruit through LC-MS/MS. Environmental Monitoring and Assessment 190: 293. 10.1007/s10661-018-6655-x
Wang, L., Huang, X., Wang, D., Chen, Y., Xu, D. and Zhou, Y., 2015. Determination of 32 sulfonylurea herbicide residues in sweet corns and green soybeans by QuEChERS-liquid chromatography-tandem mass spectrometry. Chinese Journal of Chromatography 33: 501–507. 10.3724/SP.J.1123.2014.12043
Yadav, R., Bhullar, M.S., Kaur, S., Kaur, T. and Jhala, A.J., 2017. Weed control in conventional soybean with pendimethalin followed by imazethapyr+ imazamox/quizalofop-p-ethyl. Canadian Journal of Plant Science 97: 654–664. 10.1139/CJPS-2016-0123
Yagüe, C., Bayarri, S., Lázaro, R., Conchello, P., Ariño, A. and Herrera, A., 2001. Multiresidue determination of organochlorine pesticides and polychlorinated biphenyls in milk by gas chromatography with electron-capture detection after extraction by matrix solid-phase dispersion. Journal of AOAC International 84: 1561–1568. 10.1093/jaoac/84.5.1561
Zhang, Q., Zhu, L., Wang, J., Xie, H., Wang, J., Han, Y. and Yang, J., 2013. Oxidative stress and lipid peroxidation in the earthworm Eisenia fetida induced by low doses of fomesafen. Environmental Science and Pollution Research 20: 201–208. 10.1007/s11356-012-0962-5
Zhao, Y., Wang, Y., Wang, L. and Zhang, D., 2019. Molecular identification of mung bean accessions (Vigna radiata L.) from northeast china using capillary electrophoresis with fluorescence-labeled SSR markers. Food and Energy Security 9: e182. 10.1002/fes3.182